Individual-specific edge-network analysis for disease prediction
1. 摘要
在健康不可逆转恶化之前预测疾病前状态或临界点是一项困难的任务。基于动态网络生物标志物(DNB)理论的边网络分析(ENA)通过挖掘组学数据丰富的动态和高维信息,为研究这一问题开辟了一条新的途径。尽管理论上ENA能够识别疾病进展过程中的疾病前状态,但它需要多个样本对每个个体进行这种预测,这在临床实践中通常是不可用的,从而限制了其在个性化医学中的应用。为了克服这一问题,我们提出了用DNB检测个体特异性ENA(iENA)的方法,以单样本的方式识别每个个体的疾病前状态。特别是,除了传统的疾病诊断外,iENA还可以识别出用于疾病预测的个体特异性生物标记物。为了验证该方法的有效性,应用iENA对H3N2队列的组学数据进行分析,准确地检测出每个个体流感感染的发生时间和事件的预警信号,实际达到AUC大于0.9。iENA不仅发现了新的个体特异性生物标记物,而且恢复了以往研究报道的流感感染常见生物标记物。此外,iENA还检测出具有重要边缘生物标志物的多发癌的临界期,并通过TCGA数据和其他独立数据的生存分析进一步验证了这一点。 # 2. 介绍
疾病进展一般可分为三个阶段或状态,即正常状态、病前状态(或临界点)和疾病状态。传统的分子生物标志物是根据分子数据诊断疾病状态,而不是预测疾病的预测状态。高通量技术的最新进展为研究疾病在人(或患者)体内的发生和发展提供了前所未有的机会(1,2),为准确、早期诊断个体疾病铺平了新的道路,这是精准医学的关键概念和行动(3)。
一个典型的例子是急性呼吸系统疾病的预测。对个体嫌疑人的时间转录组数据的分析揭示了许多生物学和生物医学的见解(4-6)。传统的差异基因表达分析,即传统的分子生物标志物,期望能捕获病毒感染的反应基因(7);然后基因模块分析倾向于寻找与病毒感染相关的具有特定生物学功能的相互作用基因(6,8);进一步的基因网络分析试图通过推断上游基因和下游基因之间的生物学途径来提取病毒感染的系统特征(9)。所有这些工作主要集中在利用观测数据的一阶统计量信息(如基因或蛋白质差异表达的“平均值”)诊断疾病状态。与这种传统的节点网络分析相比,最近,边缘网络分析(ENA)(10)(图1A)与动态网络生物标记(DNB)(11)相结合,通过考虑二阶统计量(如基因或蛋白质差异表达的“协方差”)来检测严重疾病恶化前的预警信号或疾病前状态(或临界点)从观测数据来看。与一般的异常检测(12-15)不同,ENA可以检测到临界转变到疾病状态之前的临界点或疾病前状态。事实上,ENA在我们早期的工作中已经显示出识别疾病前或症状前状态的能力(10),但是它需要多个样本来进行这种预测,然而,在临床实践中,这通常不是每个个体都能得到的(10,16,17)。为了克服这一问题,本文将ENA从多个样本扩展到一个样本,实现个性化诊断、预测和预后。
具体地说,我们提出了带有DNB的个体特异性ENA(iENA)来识别个体特异性生物标记物(图1B),该生物标记物应用于个体受试者(或患者)基于单个样本的疾病预测。根据DNB理论(10)的三个临界点条件,iENA可以捕获疾病动态发展过程中的高阶统计信息,用于检测疾病前状态。另一方面,ENA是建立在一个很好的随机动力学基础上的,因此它可以通过减少在发现个体特异性边缘生物标志物时的假阳性来预测个体的疾病前状态。
基于差异表达均值、差异表达方差和差异表达协方差(18,19),我们将ENA框架扩展到了iENA,并实现了对流感感染和癌症恶化等个性化疾病预测的分析。从个体受试者的H3N2队列数据来看,与传统的分子生物标志物诊断的疾病状态相比,iENA能够检测出作为每个个体流感感染临界点的预警信号或疾病前状态。特别是,(i)iENA恢复了先前工作(7)中报告的流感感染的常见生物标记物,这也比WGCNA方法(20)中的常见模块更有效;(ii)iENA根据每个受试者的时间(或空间)转录组数据和与以往疾病状态的静态预测不同,iENA采用了疾病动态预测模型DNB,成功地预测了疾病事件和时间。对于TCGA癌症数据,iENA也证明了它有能力用重要的边缘生物标记物来检测癌症的关键阶段。结果表明,iENA能同时识别乳腺癌和肝癌的临界期,TCGA数据和其他独立数据的生存分析进一步验证了该结果。
总之,iENA为基于边缘诱导数据转换的复杂疾病研究提供了强大的网络分析工具,同时也开辟了一种新的方法来预测疾病进展过程中的疾病前状态(例如,寻找癌症临界点)基于个体样本的个性化或精确医学(21)。iENA的matlab代码可以在http://sysbio.sibcb.ac.cn/cb/chenlab/software.htm。
3. 材料和方法
多样本的节点网络
生物化学主方程通常用于在分子水平上模拟生物系统的随机动力学(见补充信息A1)。对于这样一个主方程,假设用高斯分布线性化,在先前的研究(10)中,生物系统通常用一组关于分子平均矢量的方程来近似,即节点网络动力学: 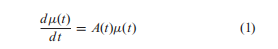 式中,μ是作为网络节点的变量的平均向量;A是底层网络的邻接矩阵,节点以浓度或数量表示分子(即μi)。显然,这组线性微分方程代表了分子网络的一阶统计信息,即平均值。它可以构造传统的网络,本文称之为节点网络,例如在分子网络中使用每个分子作为节点。
式中,μ是作为网络节点的变量的平均向量;A是底层网络的邻接矩阵,节点以浓度或数量表示分子(即μi)。显然,这组线性微分方程代表了分子网络的一阶统计信息,即平均值。它可以构造传统的网络,本文称之为节点网络,例如在分子网络中使用每个分子作为节点。
实际上,节点网络(例如分子网络)包括节点集和边集,其中节点集是分子列表,边集是分子对列表。分子对由观察数据上两个分子之间的相关性(或关联性)决定,例如基于基因表达谱的两个基因之间的皮尔逊相关系数(PCC)。众所周知,节点网络与一组样本的这种相关性可以计算为: 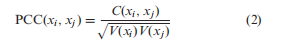 当绝对PCC非常大时,可以说xi和XJ之间有边或关联,所有边连接节点到节点网络(图1A)。注意,PCC包括两个变量之间的直接关联和间接关联,因此为了更好地表示节点网络的拓扑结构,可以采用直接关联度量,例如偏相关(22,23)或部分互信息(24),而不是等式(2)。
当绝对PCC非常大时,可以说xi和XJ之间有边或关联,所有边连接节点到节点网络(图1A)。注意,PCC包括两个变量之间的直接关联和间接关联,因此为了更好地表示节点网络的拓扑结构,可以采用直接关联度量,例如偏相关(22,23)或部分互信息(24),而不是等式(2)。 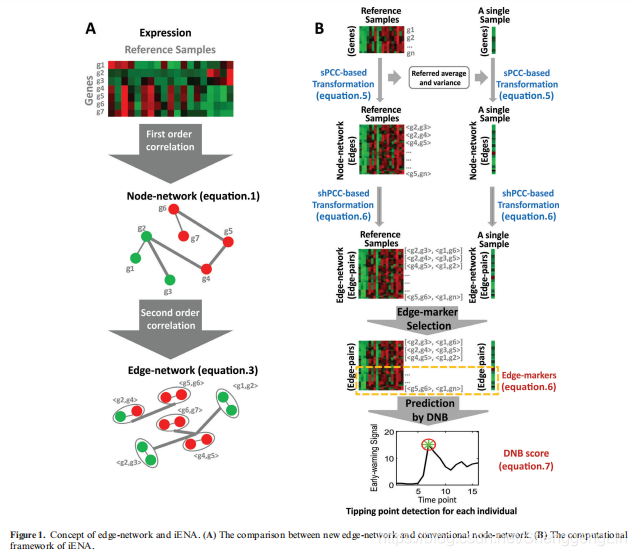 ## 多样本的边缘网络 除了方程(1)的节点网络动力学外,为了准确、完整地描述随机生物系统,还需要根据Lyapunov微分方程(见补充信息A1)在分子协方差矩阵上的另一组方程,即边网络动力学:
## 多样本的边缘网络 除了方程(1)的节点网络动力学外,为了准确、完整地描述随机生物系统,还需要根据Lyapunov微分方程(见补充信息A1)在分子协方差矩阵上的另一组方程,即边网络动力学:
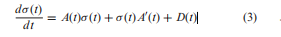 其中,σ是变量的协方差矩阵;A是原始节点网络的邻接矩阵;A'是A的转置;D是与状态无关(例如常数)的循环矩阵。显然,这是一组描述生物系统二阶统计信息的Lyapunov微分方程,即协方差。它可以利用每一对分子(即σij)或每一个分子间的相互作用作为节点来构造新的分子边缘网络。
其中,σ是变量的协方差矩阵;A是原始节点网络的邻接矩阵;A'是A的转置;D是与状态无关(例如常数)的循环矩阵。显然,这是一组描述生物系统二阶统计信息的Lyapunov微分方程,即协方差。它可以利用每一对分子(即σij)或每一个分子间的相互作用作为节点来构造新的分子边缘网络。
边缘网络中的链路由于其在两个分子对之间的关系是一个四阶统计量,这可以通过两个分子对之间的对应关系来近似推断。给定,对于四个分子,即两个分子对,相关测量计算为一组样品(10)的高阶PCC(hPCC): 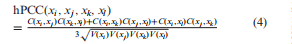 在有时间历程数据或多样本数据的情况下,可以计算出两个分子对之间的相关(hPCC),并将两个具有显著hPCC的分子对连接成一个高阶边。所有高阶边以通常的共同表达式形式将节点网络的原始边连接到新的边网络中(图1A)(10)。显然,边缘网络可以刻画生物系统的高阶矩信息。在所有分子的高斯分布假设下,随机涨落生物系统的动力学可以通过其节点网络和边网络动力学完全恢复。
在有时间历程数据或多样本数据的情况下,可以计算出两个分子对之间的相关(hPCC),并将两个具有显著hPCC的分子对连接成一个高阶边。所有高阶边以通常的共同表达式形式将节点网络的原始边连接到新的边网络中(图1A)(10)。显然,边缘网络可以刻画生物系统的高阶矩信息。在所有分子的高斯分布假设下,随机涨落生物系统的动力学可以通过其节点网络和边网络动力学完全恢复。
单样本的边缘网络
对于节点网络或边缘网络,它们的构造需要多个样本,然而,这些样本通常在临床实践中是不可用的(25)。强烈要求在单个样本(25)的基础上刻画或推断边缘网络(也包括节点网络),期望获得单个特定或样本特定的边缘网络。
以前,一个样本中PCC的量化被提议通过类相关向量(或delta PCC)来计算(26,27)。事实上,当参考样品的数量足够大时,这些测量可以减少到计算单个样品PCC(sPCC,见补充信息A1和A2): 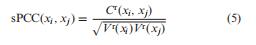 实际上,等式(5)可以看作是从单个变量xi或XJ到变量对xi-XJ的数据变换,相对于给定的参考样本组,例如从分子浓度到相关性。因此,我们可以使用该单一样本的转换数据来代替单一样本的原始数据进行进一步的分析(如差分分析或聚类分析),从而可以在单一样本的基础上揭示分子表达式的不同特征或“暗物质”。
实际上,等式(5)可以看作是从单个变量xi或XJ到变量对xi-XJ的数据变换,相对于给定的参考样本组,例如从分子浓度到相关性。因此,我们可以使用该单一样本的转换数据来代替单一样本的原始数据进行进一步的分析(如差分分析或聚类分析),从而可以在单一样本的基础上揭示分子表达式的不同特征或“暗物质”。
与PCC和sPCC类似,我们将多个样本的hPCC扩展到基于多个参考样本(见补充信息A1)的只有一个样本的shPCC,即单样本高阶PCC(shPCC)计算如下: 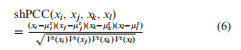 因此,类似于边缘网络的重建,我们可以从观测到的单样本数据中获得基于shPCC的个体特定边缘网络。注意,为了进一步消除等式(5)–(6)中变量之间的间接关联,可以类似地采用诸如部分相关(22,23)或部分互信息(24)之类的复杂测量。
因此,类似于边缘网络的重建,我们可以从观测到的单样本数据中获得基于shPCC的个体特定边缘网络。注意,为了进一步消除等式(5)–(6)中变量之间的间接关联,可以类似地采用诸如部分相关(22,23)或部分互信息(24)之类的复杂测量。
个体特定边缘网络分析(iENA)
最初,ENA被提议基于多个样本或群体特征(10)来研究分子关联,例如队列的风险估计。然而,越来越需要从个体的角度来分析疾病的分子机制。因此,为了解决这一网络分析的一般问题(25),在本研究中,我们提出了一个先进的框架,即iENA,基于我们提出的基于一个样本的omics数据的shPCC测量。这一新方法的细节将逐步描述(图1B)。
i) 收集数据:为了评价iENA的性能,我们从NCBI-GEO和TCGA下载了几个基因表达数据集,主要预测个体的流感病毒感染和癌症恶化。
i i)选择参考样本:为了解决单个样本(即一个时间点上一个受试者的每个样本)的平均值和方差的计算,我们需要一组参考样本(即对照样本或正常样本)。在这里,我们将从基线日期到后几点的样本设置为每个人流感感染的参考组,将正常(或早期)样本设置为癌症的参考组。实际上,任何具有类似属性的样本都可以作为参考组(参考组至少需要5个样本)。一旦确定了参考样品,在整个研究中应保持不变。
ii) 通过sPCC计算构建节点网络:当我们有参考样本时,我们可以通过PCC的单样本测量(sPCC,方程(5))来构建一个样本的共表达网络,与之前的研究(19,26,28)一致。注意,由于新sPCC值的分布不是正态分布,因此很难确定边缘相关性的直接截止值,而且我们的实验还表明,PCC的一般阈值(例如0.8或其他)在这种情况下似乎不起作用。因此,我们只关注字符串数据库(29)中的边,以减少生物上下文中的计算复杂性。需要注意的是,理论上和计算上,我们也可以从参考样本中构造sPCC的分布,而不是正态分布,而无需为此目的进行近似(截止值的统计检验),但构造此分布需要大量参考样本。此外,对于一个时间点上的每个样本,与在参考样本中观察到的相同关系相比,候选边缘/关系的as-sociations需要有很大的变化,即边缘/关系具有比其他边缘更大的标准偏差。然后,最终选择关系强且变化显著的排名靠前的边缘。这些候选边缘由传统的节点网络组成,将用作构建边缘网络的背景节点。
iii) 通过shPCC计算构造边缘网络:对于边缘网络,我们使用上述步骤的背景边缘集(基因对)作为新节点。在两个基因对之间,我们可以用shPCC(方程式(6))对每个单个样本(例如,在一个时间点对一个受试者的每个样本)的每个边缘对(即两个基因对)进行四阶单样本相关系数的估计。注意,在这个步骤中,我们实际上只计算上述步骤中预先选择的边之间的相关性,因此我们可以大大减少不必要的计算。最后,我们将得到在特定时间点对应于每个样本的边缘网络,并且每个对象在边缘网络中观察到的样本或时间序列上都有其个性化/个性化特征。
iv)通过shPCC计算构建边网络:对于边网络,我们使用来自上述步骤的一组边(基因对)作为新节点。在两个基因对之间,我们可以利用shPCC(公式(6))对每个单样本(例如,对于一个受试者在一个时间点的每个样本)对每个边缘对(即两个基因对)进行四阶单样本相关系数的估计。请注意,在这一步中,我们实际上只计算上述步骤中预先选择的边之间的相关性,因此我们可以大大减少不必要的计算。最后,我们将得到每个样本在特定时间点对应的边网络,每个被试在边网络的观测样本或时间序列上都有其个性化/个体特征。
v)识别单个边缘生物标记:与边缘选择相似,我们选择排名靠前的边缘对作为边缘生物标记,它们之间在高阶相关性方面具有很强的相关性。那些强烈相关的基因对被认为是DNB候选基因,被称为“标记”。然后,对于每个个体,边缘网络中涉及的基因对(即标记)被用作个体边缘生物标记,这些基因(即标记基因)被应用于疾病预测。
- sCI检测引爆点和DNB成员:DNB的开发是基于三个统计条件(10,16,17),识别疾病前状态或疾病进展过程中突然恶化之前的引爆点(16,30)作为一般的预警信号。最近,基于多个样本(11,31 - 34)的DNB模型及其判据(即CI: composite index)已被采用,成功识别细胞命运决定的临界点(35,36),研究免疫检查点封锁(37,38),并量化边缘生物标志物(10):
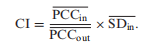 本文在单样本的基础上,通过上述二阶矩测量,进一步重新定义了DNB准则,即单样本综合指数(sCI)定义为:
本文在单样本的基础上,通过上述二阶矩测量,进一步重新定义了DNB准则,即单样本综合指数(sCI)定义为: 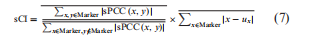 其中,分子为PCCin,为优势组基因或DNB(如一组标记基因或分子)在一个样本中绝对值表达的平均sPCC;分母为PCCout,是一个样本中优势组和其他组之间基因绝对值表达的sPCC平均值;乘数为SDin,是优势组基因或DNB表达的平均标准差。“Marker”是DNB成员的集合。上标线表示平均值。因此,当单个边缘生物标志物的sCI值大于阈值时,将提示预警信号。注意,我们可以使用式(5)的sPCC来计算式(7)的sCI,而不需要执行式(6)的shPCC。
其中,分子为PCCin,为优势组基因或DNB(如一组标记基因或分子)在一个样本中绝对值表达的平均sPCC;分母为PCCout,是一个样本中优势组和其他组之间基因绝对值表达的sPCC平均值;乘数为SDin,是优势组基因或DNB表达的平均标准差。“Marker”是DNB成员的集合。上标线表示平均值。因此,当单个边缘生物标志物的sCI值大于阈值时,将提示预警信号。注意,我们可以使用式(5)的sPCC来计算式(7)的sCI,而不需要执行式(6)的shPCC。
比较边缘生物标记:对于每个个体,我们可以使用每个单一样本中的差异基因对(即每个样本或时间点中的边缘关联)作为新的边缘生物标记来指示早期预警信号或临界点。我们将获得每个受试者或样本的边缘生物标志物sCI值,我们可以在连续的时间点或阶段观察到不同的sCI分数。因此,我们可以设置一个阈值来指示临界状态,即警告或不警告。此外,对于流感感染数据,我们还检查了每个受试者诱导的边缘生物标记,并将其与之前报道的50个基因标记和22个来自人群重点研究的基因标记进行比较(6 - 8,10)。 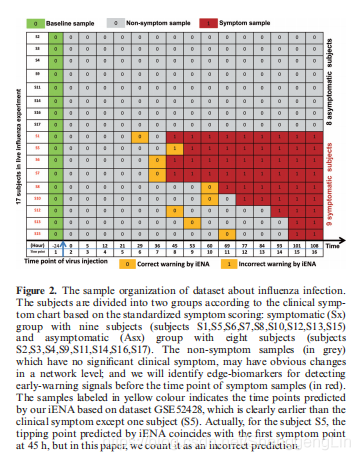
4. 结果和讨论
流感感染数据集
为了评估iENA的适用性,我们从NCBI GEO下载了两个数据集GSE30550和GSE52428(7,8)来预测受试者的实时流感感染。为了方便起见,下面主要介绍数据集GSE52428上的分析设置。
转录组数据集包含17个受H3N2/Wisconsin流感挑战的受试者(或成人)。在这样的挑战中,有9名受试者已经实际感染(即临床症状的出现),但其他8名受试者仍然保持健康(没有临床症状的出现)。收集所有受试者接种后8 h至108 h的全外周血基因表达谱并进行测定。在包括基线(受试者注射流感病毒前24小时,例如- 24 hpi)在内的16个时间点,共获得268个基因微阵列(7),这一过程与之前的研究相似(10)。
如图2所示,根据iENA,我们将受试者分为两组根据临床症状图基于标准化的症状评分(7):症状(Sx)组9个科目(科目1、5、6、7、8、10、12、13、15)和无症状(Asx)组有八个主题(主题2、3、4、9、11、14、16、17)。从基线日期(图2中的绿色部分)到后几个点的样本将作为样本的参照组;non-symptom样本(图2中灰色)没有明显的临床症状,在分子水平上可能有明显的变化,我们将确定edge-biomarkers检测预警信号症状时间点之前的样品(如图2)红色。在计算,我们五个样品包括基线数据作为参照组,即有5个参考样本可供每个主题的数据(除了13日主题错过了一次数据集GSE52428点)。然后,我们可以计算每个样本的sPCC(与参考组的均值和方差)。实验中,我们将重点放在与参考组相比变化较大的边缘(以log2(fold change) > 0.8),最终确定了每个时间点的前1000强关系。然后,使用这些预先选择边缘为背景的“节点”建设移动网络,并捕获的重要信号峰值edge-biomarkers跨多个时间点,从59基因最终获得相关edge-biomarkers Sx组(称为Sx基因)比215年候选Asx组。这些基因是候选的DNB成员。 ## iENA鉴定的流感感染的边缘生物标志物与文献报道一致 iENA基于来自数据集GSE52428的每个样本,预测了每个受试者的引爆点,如图2所示(即标记为黄色的样本),除了一个受试者(S5),这些样本明显早于临床症状(S5)。实际上,对于受试者S5, iENA预测的引爆点与第一个症状点在45小时一致,但在本文中,我们认为这是一个不正确的预测。iENA恢复了以往工作中报道的流感感染的常见生物标志物(10),证明了iENA在疾病生物标志物发现方面的有效性(见补充资料A3)。如图3A所示,在Sx个体中检测到的标记基因与在Asx个体中检测到的标记基因有很大差异;这就是为什么sx特异性边缘标记可以用来预测症状的出现(或疾病的发生)。如图3B所示,我们最终的边缘生物标记与我们之前工作中报道的22个基因标记(10)和原始工作中报道的50个基因标记(6 - 8,10)有显著重叠。这意味着我们的新方法可以有效地找到与流感感染有关的关键基因,并发现许多新的与疾病相关的基因。
特别地,我们评估了edge-network在个体上回收之前报道的标记的效率,并使用重叠比(即每个个体的重叠基因与所有样本上检测到的全部标记基因的比值)来评估这种效率。如fig . 3C所示,Sx个体的报道基因数接近10%,而Asx个体的报道基因数<5%。这一事实意味着在Sx个体上观察流感感染的变化比在Asx个体上更有可能,并且边缘网络在预测疾病方面是有效的。此外,我们还评估了节点网络的相似效率(即由背景边构造)。如图3D所示,Sx和Asx个体的重叠率均<1%,且无显著性差异,再次支持了边网络比节点网络在iENA中更有效的观点。
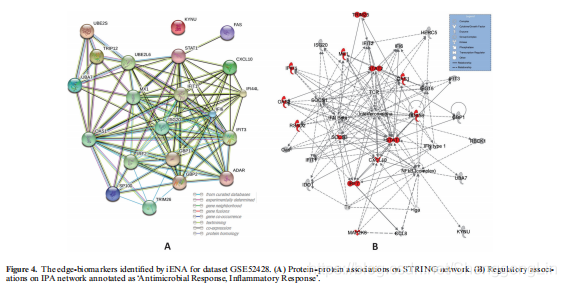
接下来,我们的边缘生物标记物(即Sx特异性边缘生物标记物)在字符串网络上有许多紧密的关联(图4A),这表明边缘网络实际上可以检测相互作用的基因对。这些标记基因在许多与病毒相关的KEGG途径,如NOD样受体信号途径、单纯疱疹病毒感染、甲型流感、丙型肝炎、胞浆DNA传感途径和麻疹等方面都有丰富的表达。对于甲型流感途径,我们的标记基因(如图4B中的红色)广泛分布在IPA注释的网络上,称为“抗菌反应,炎症反应”,据报道,这与甲型流感病毒感染的早期免疫反应有关(39)。这有力地支持了我们的生物标记物能够在症状出现之前检测到感染的预警信号(即症状前状态或临界点的预测)。 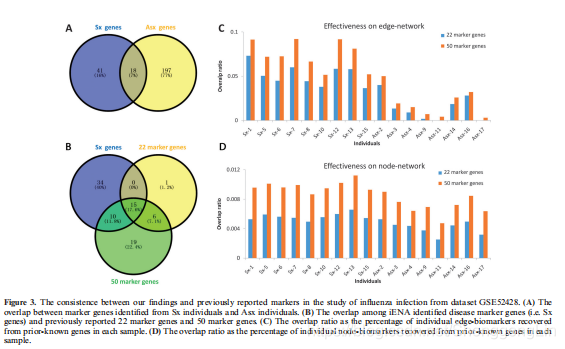
5. 结论
我们提出了一个新的框架,即基于DNB的iENA,通过从生物数据中以一个样本的方式提取高阶统计量和动态信息来识别疾病进展过程中的疾病前状态或临界点。本文以iENA为例,主要分析了17例有流感感染危险的健康成人的两个时程数据集和两个TCGA癌症数据集。iENA分析的流感感染数据结果表明:(1)9例Sx受试者除1例外,在临床诊断前均能正确地发现症状的预警信号,并检测出其具有边缘生物标志物(即DNB)的临界时间点(即临界点);(ii)边缘生物标记物与疾病的进展和发展(如病毒感染)显著相关;和(iii)边缘生物标记物能够同时预测感染的发生和时间。此外,我们应用iENA分析TCGA癌症蛋白质组数据(乳腺癌和肝癌),这也揭示了癌症发展的关键阶段,并提供了与生存相关的蛋白质特征。这些结果证明了用DNB进行iENA分析在疾病研究中的有效性,这使得它在临床应用中不需要多个样本,这是针对不同类型的组学数据进行个性化医疗的有用工具(25)。
事实上,iENA是ENA的进一步发展,从预测多个样本的共同危险因素到预测单个样本上的个体特异性生物标记物。这种新方法实际上是利用个体样本来预测疾病状态,通过探索基于差分表达式、方差和协方差分析的差分边缘网络。尽管我们的评估表明,iENA在参数设置上是稳健的(见补充信息A3),但在参考样本有限的情况下,它需要仔细的质量控制以减少实验误差。在这项工作中,我们关注的是组学数据而不是低吞吐量数据,因此,目前尚不清楚iENA是否可以直接应用于临床面板分析。如何优选边缘生物标志物作为DNB成员也是未来的研究课题。iENA将是迈向精确医学的重要一步,尤其是其预测包括癌症在内的疾病临界点或临界转变的能力也是转化医学领域的一个重要课题。除医学外,iENA还可直接应用于许多生物学过程的临界点和关键调控因子的研究,如细胞分化或分子进化。
原论文名称:Individual-specific edge-network analysis for disease prediction