Toward Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-Based Convolutional Encoders(1)
0. 摘要
根据最近神经网络在药物设计和灵敏度预测方面的进展,我们提出了一种新的模型,利用基于多模态注意机制的卷积编码器对抗癌化合物灵敏度进行可解释预测。我们的模型基于药物敏感性的三个关键数据:化合物结构(SMILES),肿瘤的基因表达谱,以及来自蛋白质-蛋白质相互作用网络的细胞内相互作用的先验知识。我们证明,我们的多尺度卷积注意力编码器显著优于基于Morgan指纹训练的基线模型和基于SMILES的编码器选择,以及先前报道的最先进的多模态药物敏感性预测。此外,通过对注意权重的深入分析,证明了本文方法的可解释性。我们发现参与基因显著地丰富了凋亡过程,并且药物注意与标准化学结构相似指数密切相关。最后,我们报告了一个案例研究的两个受体酪氨酸激酶(RTK)抑制剂作用于白血病细胞系,展示了模型的能力。 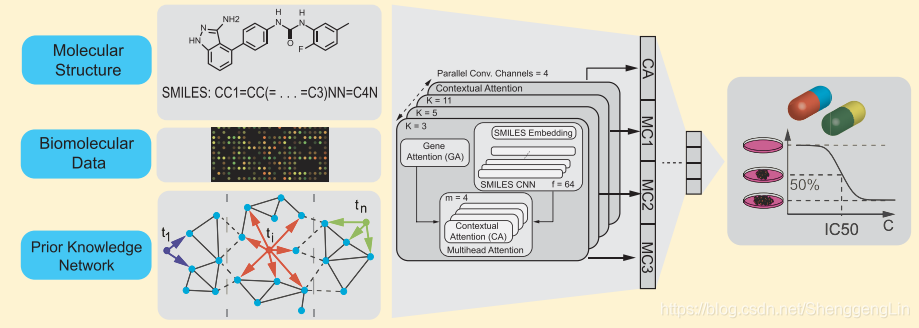
1. 介绍
发现具有理想疗效的新型化合物和改进现有疗法是制药行业的关键瓶颈。特别是抗癌化合物,在药物研发中所占份额最大,占2018年全球研发计划药物总数的34%以上(15 267种药物中有5212种)。尽管近年来科学技术取得了巨大的进步,但在抗癌药物的发现中,偶然性仍然扮演着重要的角色。另一方面,有强有力的证据表明,抗癌治疗的反应高度依赖于肿瘤基因组和转录组组成,这导致了患者对抗癌药物的临床反应的异质性。这种不同的临床反应带来了个性化(或精准)癌症治疗的前景,从患者的肿瘤图谱中获得的分子生物标记物,例如特定基因的表达,可以用于选择个性化治疗。
这些挑战突出了制药和医疗保健行业对多模态定量方法的需求,这些方法可以共同利用不同的数据来源,以表征化合物的分子结构、生物样品的遗传和表观遗传改变以及药物反应之间的联系。在这项工作中,我们提出了一种多模态方法,使我们能够应对上述挑战。
2. 方法
2.1 数据
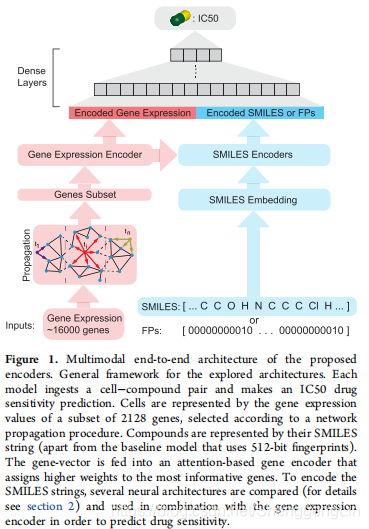
在整个研究过程中,我们使用了公开可用的癌症药物敏感性基因组学(GDSC)数据库中的药物敏感性数据。该数据库包含了1000多种具有基因图谱的人类泛癌细胞系的筛选结果,这些细胞系含有广泛的抗癌化合物(化疗药物和靶向治疗药物)。药敏值在对数刻度上以半数最大抑制浓度(IC50)表示。从收集到的canonical SMILES中,我们使用RDKit (512-bit,半径为2)获得了Morgan指纹。由于SMILES语言的性质,大多数分子都可以通过不同的SMILES字符串有效地表示(例如,C(=o)=o和O=C=O都表示二氧化碳)。利用这一特性,Bjerrum提出了一种有效的数据增强策略。我们选择用每个细胞的转录组谱来表示它们,因为已经证明,与其他组学数据相比,转录组数据更能预测药物敏感性。因此,所有可用的RMA规范化基因表达数据都从GDSC数据库中检索出来,总共得到985个细胞系的转录组谱。
2.2 网络传播
由于GDSC 985个细胞系中的每一个最初都是由17 737个基因的表达水平来表示的,因此信息特征减少是必不可少的,因为我们发现处理高维原始数据在计算上是困难的。为此,我们利用STRING蛋白-蛋白相互作用(PPI)网络(一个综合PPI数据库,包含多个数据源的相互作用)进行网络传播,留下2128个基因子集。我们采用的加权和网络传播方案包括以下步骤:我们首先给报告的药物靶基因分配一个高权重(W = 1),而给所有其他基因分配一个非常小的正权重(ε = 1 × 10−5)。此后,初始化的权值在STRING上传播。这个过程是为了将分子相互作用的先验知识整合到我们的加权方案中,并模拟给药后细胞内扰动的传播。设初始权值为W0,STRING网络S = (P, E, A),其中P为网络的蛋白质顶点,E为蛋白质之间的边,A为加权邻接矩阵。平滑权值由传播函数的迭代解确定 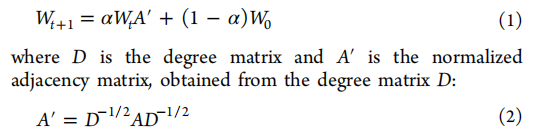 扩散调节参数α(0≤α≤1)定义了先验知识权重在网络中的扩散程度。在这项工作中,我们使用了α = 0.7,正如文献中推荐的STRING网络采用e = (Wt+1−Wt)<1 × 10−6的收敛规则,对每种药物迭代求解公式1,并利用所得权重分布确定每种药物排名前20位的基因。通过为每种药物选择前20个基因,就有可能编制出一个具有交互信息的基因子集(共2128个基因)。在输入我们的模型之前,这个基因子集被用来描述数据集中的每个细胞系。每种药物的选择仅限于前20个基因,以保证在拓扑意识和描述生物分子剖面的特征数量之间进行权衡。然后,我们对所有筛选的细胞株和药物进行配对,生成细胞-药物配对和相关的IC50药物反应的泛药物数据集。由于GDSC数据库中的缺失值,将985个细胞系与208种药物配对产生了175 603对,在SMILES增强后可以增加到超过550万个数据点。
扩散调节参数α(0≤α≤1)定义了先验知识权重在网络中的扩散程度。在这项工作中,我们使用了α = 0.7,正如文献中推荐的STRING网络采用e = (Wt+1−Wt)<1 × 10−6的收敛规则,对每种药物迭代求解公式1,并利用所得权重分布确定每种药物排名前20位的基因。通过为每种药物选择前20个基因,就有可能编制出一个具有交互信息的基因子集(共2128个基因)。在输入我们的模型之前,这个基因子集被用来描述数据集中的每个细胞系。每种药物的选择仅限于前20个基因,以保证在拓扑意识和描述生物分子剖面的特征数量之间进行权衡。然后,我们对所有筛选的细胞株和药物进行配对,生成细胞-药物配对和相关的IC50药物反应的泛药物数据集。由于GDSC数据库中的缺失值,将985个细胞系与208种药物配对产生了175 603对,在SMILES增强后可以增加到超过550万个数据点。
2.3 模型架构
 DNN。基线模型是一个六层的DNN,有[512,256,128,64,32,16]单元和一个激活函数。基线模型的超参数优化通过交叉验证方案实现(见2.4节),其中512位摩根指纹和基因表达谱(使用2.1节中描述的网络传播方法得到)被连接到一起作为模型第一层的输入。
DNN。基线模型是一个六层的DNN,有[512,256,128,64,32,16]单元和一个激活函数。基线模型的超参数优化通过交叉验证方案实现(见2.4节),其中512位摩根指纹和基因表达谱(使用2.1节中描述的网络传播方法得到)被连接到一起作为模型第一层的输入。
SMILES编码器的共性。为了研究哪种模型架构能够最好地学习化合物的分子信息,我们探索了各种SMILES编码器。raw SMILES字符串使用Schwaller等人的正则表达式被标记为单个原子,例如,四氧化二氮的SMILES字符串(N+(N+-[O−])[O−])由26个字符组成,但被分解为14个实体(N+(N+ [O−])[O−])。这确保了分子的小功能单元(如[NH]或[N+])被表示为模型中的单个实体。得到的原子序列被零填充,表示为E = {e1,…,
eT},对于每个字典token,学习到的嵌入向量ei∈RH(见图2A)。通过网络传播选择出的基因子集来表示每一个细胞系,将其输入到基因注意力编码器(见图2B)。一个与输入信息维度相同的单一softmax密度层会在基因上产生一个注意力权重分布,并在点积中对它们进行过滤,确保信息最丰富的基因得到更高的权重以供进一步处理。由此得到的基因注意力权重使模型具有可解释性,因为它们识别了驱动每个细胞系敏感性预测的基因。该体系结构也被用于深度基线模型,但由于性能较差而被丢弃。所有的SMILES编码器之后都是一组密集的层(如图1所示),使用dropout (pdrop = 0.5)进行正则化,使用sigmoid激活函数。回归是由一个线性激活(而不是sigmoid)的单个神经元完成的,以避免将值限制在0到1之间,从而阻碍网络的学习过程。 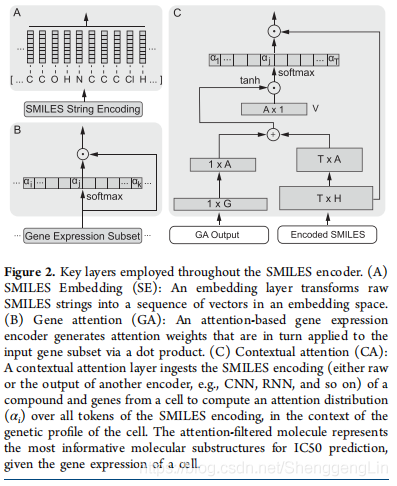 RNN传统上是序列编码的首选方法。为了研究其编码SMILES的有效性,我们采用了带有门控回归单元(GRUs)的双层双向递归神经网络(bRNN)。将前向和后向GRU-RNN的最终状态连接并反馈给稠密层进行IC50预测。
RNN传统上是序列编码的首选方法。为了研究其编码SMILES的有效性,我们采用了带有门控回归单元(GRUs)的双层双向递归神经网络(bRNN)。将前向和后向GRU-RNN的最终状态连接并反馈给稠密层进行IC50预测。
Stacked 卷积编码器(SCNN)。接下来,我们使用了一个具有四层Stackedd卷积和sigmoid激活函数的编码器。第一层的2D卷积核分解了嵌入向量的隐藏维数,随后的1D卷积提取了分子不同部分之间越来越长的依赖关系。因此,与bRNN类似,SCNN微笑编码器的任何输出神经元都整合了整个分子的信息。
Self-Attention (SA)。我们研究了几个利用神经网络注意力机制的编码器,最初是由Bahdanau等人提出的。可解释性在医疗保健和药物发现中至关重要。因此,神经注意力机制在我们的模型中是核心的,因为它们使我们能够在潜在的生物和化学过程的背景下解释和解释观察到的结果。我们的第一个注意配置是一个自注意(SA)机制,它改编自文档分类,用于编码微笑字符串。每个原子标记计算微笑的注意权重αi为  Contextual-Attention (CA)。此外,我们还设计了一种上下文注意(CA)机制,利用基因表达子集G作为上下文(图2C)。注意权重αi由如下公式确定:
Contextual-Attention (CA)。此外,我们还设计了一种上下文注意(CA)机制,利用基因表达子集G作为上下文(图2C)。注意权重αi由如下公式确定: 
 多尺度卷积注意力机制(MCA)。在最简单的形式中,SA和CA模型的注意机制直接作用于嵌入,忽略了位置信息和长期依赖性。有趣的是,注意力模型的表现仍然优于整合了整个分子信息的bRNN和SCNN。为了结合基于注意的模型的优点,即可解释性和序列编码器提取局部和长期依赖的能力,我们设计了多尺度卷积注意(MCA)编码器,如图3所示。使用MCA,用三个单独的通道分析化合物的SMILES字符串,每个通道将SMILES嵌入与一组尺寸为[H, 3], [H, 5]和[H, 11]的f核卷积和ReLU激活。药物的功效可能主要与附在受体结合位点的特定分子亚结构的出现有关。MCA被设计用来利用其可变的内核大小来捕获各种大小的子结构。例如,一个特殊的核可以检测到类固醇结构,这在抗癌分子中很常见。在多尺度卷积之后,每个通道的结果特征图被输入到上下文注意层,该层将过滤后的基因作为上下文接收。与Vaswani等人类似,我们为每个通道使用了m =
4个上下文注意层,以便让模型共同参与分子的几个部分。多头注意方法,抵消了softmax过滤掉绝大多数顺序步骤的趋势。在第四个通道中,跳过卷积,直接将原始SMILES嵌入到并行CA层。这4m层的输出在被输入到密集的前馈层之前被连接起来。
多尺度卷积注意力机制(MCA)。在最简单的形式中,SA和CA模型的注意机制直接作用于嵌入,忽略了位置信息和长期依赖性。有趣的是,注意力模型的表现仍然优于整合了整个分子信息的bRNN和SCNN。为了结合基于注意的模型的优点,即可解释性和序列编码器提取局部和长期依赖的能力,我们设计了多尺度卷积注意(MCA)编码器,如图3所示。使用MCA,用三个单独的通道分析化合物的SMILES字符串,每个通道将SMILES嵌入与一组尺寸为[H, 3], [H, 5]和[H, 11]的f核卷积和ReLU激活。药物的功效可能主要与附在受体结合位点的特定分子亚结构的出现有关。MCA被设计用来利用其可变的内核大小来捕获各种大小的子结构。例如,一个特殊的核可以检测到类固醇结构,这在抗癌分子中很常见。在多尺度卷积之后,每个通道的结果特征图被输入到上下文注意层,该层将过滤后的基因作为上下文接收。与Vaswani等人类似,我们为每个通道使用了m =
4个上下文注意层,以便让模型共同参与分子的几个部分。多头注意方法,抵消了softmax过滤掉绝大多数顺序步骤的趋势。在第四个通道中,跳过卷积,直接将原始SMILES嵌入到并行CA层。这4m层的输出在被输入到密集的前馈层之前被连接起来。 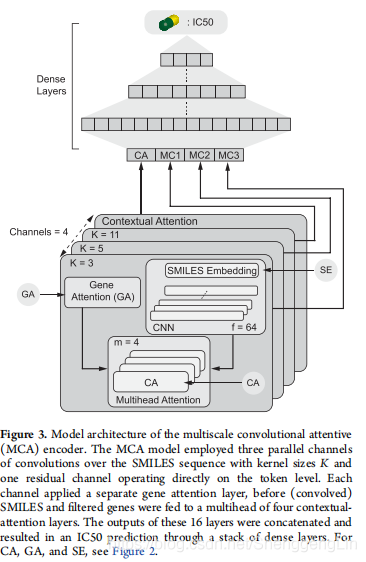 ## 2.4 模型评估
## 2.4 模型评估
为了对不同的架构进行基准测试,我们采用了严格的数据分割方法,以确保验证或测试数据集中的细胞系和复合结构在验证或测试之前都没有被我们的模型看到。在我们的数据分割策略中,从GDSC数据库中提取了总数为208个化合物和985个细胞系的10%子集,作为一个不可见的测试数据集来评估训练后的模型。剩下的90%的化合物和细胞系用25折交叉验证方案进行模型训练和验证。在每折中,4%的药物和4%的细胞系被分离并用于生成验证数据集,剩余的药物和细胞株被配对并输入模型进行训练。在实践中,这种策略剥夺了模型的很大一部分样本,这些样本没有被分类到任何训练、验证或测试数据中。
为了将我们的模型与先前选择了不那么严格的数据分割策略的工作进行比较,我们采用了类似的策略。这种分割包括一个标准的5折交叉验证方案,其中10%的配对(来自985个细胞系和208种药物的175 603个药物对)被留出用于测试。
将训练数据的IC50值归一化为[0,1],对验证和测试数据进行相同的转换。对训练集中的基因表达值进行标准化,并对验证集和测试集中的基因表达进行相同的转化。
2.5 训练过程
所有描述的体系结构都是在TensorFlow 1.10中实现的,MSE损失函数由Adam (β1 = 0.9, β2 = 0.999, ε = 1 ×10−8)优化。所有SMILES编码器采用H = 16的嵌入维数。SA和CA模型的注意维度设置为A = 256, MCA模型设置为A = f = 64。在所有模型的最后密集层中,我们采用dropout (pdrop = 0.5)、批规范化和sigmoid激活。在配备POWER8处理器和NVIDIA Tesla P100的集群上,对所有模型进行了批大小为2048的训练,最大步长为500k。 # 3. 结果 结果证明模型好。 # 4. 讨论 我们提出了一种基于注意力的多模态神经网络方法,用于解释药物敏感性预测,该方法结合了(1)药物化合物的SMILES字符串编码,(2)癌细胞的转录组学,以及(3)纳入PPI网络的细胞内相互作用。在对SMILES序列编码器的广泛比较研究中,我们证明了使用药物化合物的raw SMILES串,我们能够超越使用Morgan指纹的基线模型所达到的预测性能。此外,我们还发现基于注意的SMILES编码架构,特别是新提出的MCA,在产生可验证可解释的结果时表现最好。为了进一步提高我们模型的可解释性,我们设计了一种作用于遗传图谱的基因注意机制,并将重点放在IC50预测中信息最丰富的基因上。通过对GDSC中包含的所有细胞系进行通路富集分析,我们验证了基因注意权重的正确性,并发现凋亡过程显著富集。在对白血病细胞系的个案研究中,我们展示了我们的模型如何能够专注于相关化合物的结构元素,并考虑与感兴趣的疾病相关的基因。通过在STRING PPI网络上传播技术,我们的模型探索了2128个信息最丰富的基因,而不是全部17 737个基因。相反,利用完整的基因集会使模型训练在计算上变得难以处理;但是,没有忽略大多数基因的其他特征约简技术,如为所有相关路径获得单样本签名得分,这里还没有探索。我们基于基因的方法的明显好处是基因注意力机制,而纯粹基于通路活动分数的方法将忽略这一机制。然而,扩展我们的模型,增加通路得分的输入通道和相关的通路注意机制,可以极大地补充肿瘤细胞的表现。
我们设想我们的以注意力为基础的方法将在个性化医疗和新的抗癌药物发现中发挥巨大的作用,在这些领域,药物敏感性的可解释预测是至关重要的。这开启了一种情景,在癌症精准医学中,个性化治疗可以成为患者护理的具体选择。
原论文名称:Toward Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-Based Convolutional Encoders